Invited state-of-the-art review
Lipids and lipoproteins
26. jun. 2023
16 min.
Abstract
Keypoints
Cardiovascular disease is the number one cause of global mortality [1]. Ischaemic heart disease in particular is often caused by atherosclerosis, leading to both gradual narrowing of cardiac blood vessels and acute thrombotic events. Prevention is targeted towards lifestyle factors such as smoking, diet and physical exercise, as well as blood pressure, diabetes and blood lipids. The aim of this review is to provide a brief up-to-date overview of blood lipids and lipoproteins and their involvement in atherosclerosis.
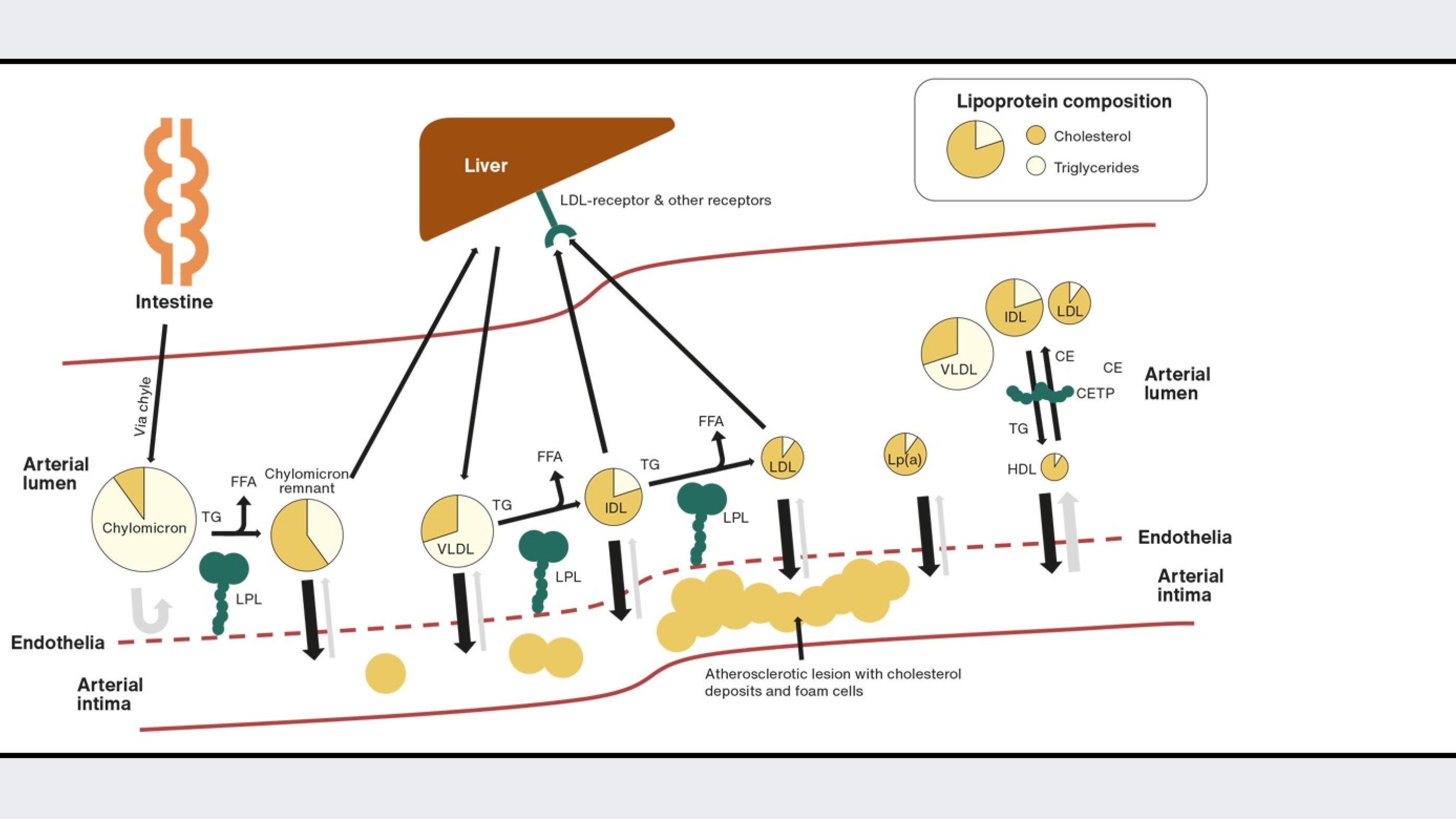
Lipids are essential for human physiology. Fatty acids, which are stored and transported as triglycerides, are used as energy, and cholesterol is essential as a structural component in cell membranes and as a precursor for hormones, vitamin D and bile acids. After a meal, lipids are emulsified by bile acids in the intestinal lumen, allowing lipases to hydrolyse triglycerides into free fatty acids and monoacylglycerols and cholesterol esters into free cholesterol (Figure 1). After entering the enterocyte, free fatty acids and cholesterol are resynthesised into triglycerides and cholesterol esters and are assembled into large apolipoprotein B-48-containing chylomicrons and secreted into the chyle from where it reaches the blood circulation. Triglyceride-rich chylomicrons are rapidly metabolised, delivering triglycerides to muscle and adipose tissues. Here, lipoprotein lipase (LPL) at the endothelial surface hydrolyses the triglycerides into free fatty acids, clearing the chylomicron remnants from the circulation via the liver. In the liver, cholesterol can be synthesised de novo with HMG-CoA reductase as the rate-limiting enzyme. The liver assembles triglycerides and cholesterol esters into apolipoprotein B-100, containing very low-density lipoproteins (VLDL), which are then secreted into the blood. As for chylomicrons, the triglycerides in VLDL are hydrolysed by LPL at the endothelia in muscle and adipose tissues. This process transforms the VLDL particles into intermediate-density lipoprotein (IDL) particles. The IDL particles can either be cleared by the liver or transformed into low-density lipoproteins (LDL) by further LPL hydrolysis of triglycerides [2]. In turn, LDL particles are cleared by LDL receptors in the liver and peripheral tissues [3].
Cholesterol can also be transported from peripheral tissues to the liver by high-density lipoproteins (HDL) in what is termed reverse cholesterol transport. The HDL particle is formed when circulating apolipoprotein A-1, produced in the liver or intestine, is lipidated with cholesterol and phospholipids from the liver, intestine and other tissues. The cholesterol in the nascent HDL particle is then esterified by lecithin-cholesterol acyltransferase bound to the HDL particle, generating spherical mature HDL particles. In the circulation, the HDL particle can exchange its cholesterol esters with triglycerides from apolipoprotein-B-containing lipoproteins, chylomicrons, VLDL and LDL, mediated by the cholesterol-ester transfer protein (CETP) [4]. In the liver, HDL delivers cholesterol to hepatocytes from where it can be secreted via bile to the intestine.
Finally, without having a well-described role in lipid metabolism, lipoprotein(a) needs to be described because of its role in cardiovascular disease. Lipoprotein(a) is a lipoprotein consisting of an apolipoprotein-B-100-containing LDL-like particle with an additional apolipoprotein, apolipoprotein(a), attached to apolipoprotein B-100. Apolipoprotein(a) is produced and secreted from hepatocytes. However, the exact place of assembly of the lipoprotein(a) particle is not well understood and the same is true for the catabolism of the particle. What is known is that apolipoprotein(a) consists of so-called kringle domains with homology to plasminogen of which some are present in numerous repeats. The number of these are determined by the number of genetic repeats in LPA, leading to the varying size of apolipoprotein(a). Apolipoprotein(a) with a low number of kringle repeats is associated with a high plasma concentration of lipoprotein(a) and vice versa, making plasma levels of lipoprotein(a) 90% genetically determined [5].
In 1913, Nikolai Anitschkow first described the pathological process from cholesterol ingestion to cholesterol deposition in the arterial wall in rabbits and thereby identified the process of atherosclerosis [6]. Adolf Windaus observed the abundance of cholesterol in aortas with atherosclerotic plaques, placing cholesterol in the context of atherosclerosis prior to Anitschkow’s findings by several years. The atherosclerotic process is initiated by accumulation of lipoproteins and lipids in the arterial intima. LDL, lipoprotein(a) and remnant lipoproteins can penetrate the arterial endothelium and be retained in the intima by binding to proteoglycans in the extracellular matrix. This can trigger local inflammation and activation of the endothelium, which then recruits monocytes to the intima. In the intima, monocytes differentiate into macrophages and ingest the lipoproteins, in turn creating cholesterol-rich foam cells [7]. The atherosclerotic process continues its progression with migration of smooth muscle cells to the intima from the arterial media layer. This process produces collagen and other extracellular matrix molecules, building the fibrous cap of the atherosclerotic lesion [7]. In the advanced lesion, foam cells and other cells die spilling out their cholesterol content, and cholesterol crystals develop creating a necrotic core. Finally, the fibrous cap may rupture as a result of the underlying necrotic core. This initiates thrombus formation as circuiting coagulation components come into contact with tissue factors from the atherosclerotic lesion, restricting blood flow and oxygen delivery to downstream tissues [3]. Whereas inflammation plays a critical role in atherosclerotic progression, cholesterol is the sine qua non of this disease. However, determining which lipoprotein cholesterol fractions are involved and how to target these in treatment of the disease is an ongoing area of development.
Low-density lipoprotein cholesterol
LDL cholesterol has long been a firmly established causal factor in the development of atherosclerosis and cardiovascular disease (Table 1). A comprehensive review of the evidence is provided by Ference et al. in the European Heart Journal 2017 [8]. In the clinic, LDL cholesterol is often calculated from total cholesterol, HDL cholesterol and triglycerides using the Friedewald equation. However, new equations have recently been developed, offering better estimations of LDL cholesterol, especially in patients with low LDL cholesterol and/or high triglycerides [9, 10]. Direct measurement of LDL cholesterol is also available and is warranted in patients with high triglycerides. Statins, the main lipid-lowering drug class reduce LDL cholesterol by reducing HMG CoA reductase activity. Thus, the endogenous production of cholesterol in the liver leads to a compensatory increase in LDL receptor expression at the surface of the hepatocyte. In some patients, the desired lowering of LDL cholesterol cannot be achieved with statins alone. For these patients, the new add-on treatment options recommended in current guidelines are ezetimibe, which decreases intestinal cholesterol uptake by inhibiting the Niemann-Pick C1-like 1 protein, and proprotein convertase subtilisin/kexin type 9 inhibitors, which reduce LDL receptor degradation[11, 12]. In patients who cannot or refuse to take a statin, bempedoic acid is an alternative though it has a lesser cholesterol-lowering effect than statins [13].
Remnant cholesterol
Remnant cholesterol, the cholesterol in VLDLs and IDLs, is the centre of increasing interest. As remnant cholesterol and triglycerides are carried in the same lipoproteins, they are closely correlated. Genetic evidence suggests that beyond contributing to atherosclerosis, remnant lipoproteins cause low-grade inflammation [14]. Several studies have shown a positive association between elevated levels of remnant cholesterol, triglycerides and the risk of cardiovascular disease [15-17]. In high-risk patients with diabetes or elevated cholesterol and in patients receiving statin-therapy, remnant cholesterol indeed is associated with incident cardiovascular disease, whereas LDL cholesterol, in some studies, is not [18-20]. This points to an emerging role of remnant cholesterol in residual risk of cardiovascular disease. Whereas triglycerides from remnant lipoproteins can be hydrolysed and serve as energy supply, the cholesterol may accumulate in the intima, leading to atherosclerosis. No trials have targeted remnant cholesterol.
However, randomised controlled trials focused on reducing triglycerides have so far shown contradictory results. While the STRENGTH [21] and REDUCE-IT [22] trials, which used different formulations of omega-3 fatty acids, both succeeded in reducing triglycerides by around 20%, a lower risk of cardiovascular events compared with placebo was observed only in the REDUCE-IT trial. This effect was possibly attributable to a 10% increase in LDL cholesterol in the placebo group receiving mineral oil. In the PROMINENT trial [23], pemafibrate, a selective peroxisome proliferator-activated receptor α modulator, reduced triglycerides but also increased LDL cholesterol and apolipoprotein B. However, pemafibrate did not reduce the incidence of cardiovascular events. Despite this, a causal role of remnant cholesterol in cardiovascular disease has gained much support from Mendelian randomisation studies in which genetic variants in several genes associated with elevated remnant cholesterol and triglycerides were also associated with an increased risk of cardiovascular disease [16, 24-26]. Mendelian randomisation studies also identified ANGPTL3 [27] and APOC3 [28, 29] as new drug targets, leading to the development of evanicumab, vupanorsen [30, 31], volanesorsen [32] and olezarsen [33]. These drugs all potently reduce remnant cholesterol and triglycerides, but they have not yet been tested for their ability to reduce cardiovascular events.
Non-high-density lipoprotein cholesterol
Both LDL and remnant lipoproteins are thought to contribute to atherosclerosis as described above. This has promoted the inclusion of non-HDL cholesterol, the sum of all cholesterol in atherogenic lipoproteins, in the standard lipid profile. Together with apolipoprotein B, representing the total number of atherogenic lipoproteins, non-HDL cholesterol is generally considered an alternative to LDL cholesterol levels in patients with hypertriglyceridemia or diabetes [11, 34, 35]. Whereas apolipoprotein B measurement is not widely available in clinical laboratories, non-HDL cholesterol may be calculated and reported from the standard lipid profile without extra costs. Also, non-HDL cholesterol has been incorporated into the SCORE2 [36] and SCORE2-OP [37] charts recommended by the European Society of Cardiology [11] to identify cardiovascular risk.
Lipoprotein(a)
Lipoprotein(a), like LDL and remnant lipoproteins, can infiltrate the arterial intima. There, its atherogenic potential is both related to its cholesterol content and proinflammatory and prothrombotic properties. Strong evidence from epidemiological and genetic studies points at lipoprotein(a) as a causal factor in the development of cardiovascular disease [38-40]. The role of lipoprotein(a) in the clinical setting has been put off by at least two issues; how and when to measure, and how to deal with findings of elevated plasma levels. For measuring lipoprotein(a), standardisation of assays has improved, and assays are now available for automated platforms [41]. Furthermore, guidelines now recommend lipoprotein(a) measurement at least once in all adults [34, 35] and two potent lipoprotein(a)-reducing drugs are in clinical trial [42, 43] (NCT04023552 and NCT05581303).
High-density lipoprotein cholesterol
A low level of HDL cholesterol is a strong prospective marker of high cardiovascular risk. This was established in the Framingham Study [44] and has subsequently been confirmed in various other populations. Even so, clinical trials of drugs that increase HDL cholesterol, extended-release niacin and CETP inhibitors have generally not succeeded in reducing the risk of cardiovascular events [45-48]. Only anacetrapib [49] which increases HDL cholesterol by more than 100% but also decreases LDL and lipoprotein(a), succeeded in reducing the risk of cardiovascular disease events by a meagre 9%. Results from Mendelian randomisation studies have been conflicting and have generally failed to establish a link between genetically elevated levels of HDL cholesterol and a reduced risk of cardiovascular disease. Together these findings have shifted the focus from HDL cholesterol per se as a beneficial causal factor in reducing cardiovascular disease to remnant cholesterol and triglycerides as the strong association between HDL cholesterol and cardiovascular risk may be explained via an equally strong inverse association with remnant cholesterol and triglycerides. In fact, low HDL cholesterol may be used as a long-term marker of elevated triglycerides and remnant cholesterol, just like elevated HbA1c is a long-term marker of plasma glucose [50].
Lipids are an essential part of human physiology but also cause atherosclerosis and cardiovascular disease when elevated. While LDL cholesterol is the primary lipid in risk assessment and the primary target in cardiovascular disease prevention, other lipoproteins are gaining increasing attention. Genetic evidence suggests that remnant cholesterol, triglycerides and lipoprotein(a) are important causal lipid fractions in atherosclerosis development and emerging therapeutic targets that may reduce residual cardiovascular risk.
Correspondence Anders Berg Wulff. E-mail: anders.berg.wulff.01@regionh.dk
Accepted 22 May 2023
Conflicts of interest Potential conflicts of interest have been declared. Disclosure forms provided by the authors are available with the article at ugeskriftet.dk/dmj
Cite this as Dan Med J 2023;70(7):A03230185